Morpheus
Prediction of Transcription Factors Binding Sites based on Position Weight Matrix
User guide
SEQUENCES
Sequences must be in standard FASTA format. In order to facilitate data visualization and identification we suggest to use as short as possible sequence names and to pay attention to duplicated names. Anyway fasta file is processed so a number between parenthesis are added if a duplicated name is found.
Not allowed characters in sequence name :
-
Avoid the use of "-" in sequence name as is used as mark for genomic information (see bellow). If you use it in a wrong way, name can be truncated incorrectly by some programs when writing results, what can difficult correct identification of sequence when interpreting results.
-
Characters "\(:)/?*|<>" can't be used in sequences name as several programs use it for creating individual profiles and data results files. By default, fasta file is processed so those characters are erased before creating file (white spaces are also substituted by "-").
Example of Fasta Format :
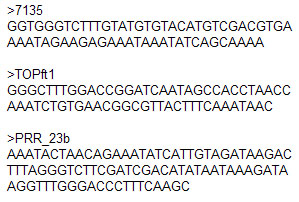
Unambiguous sequences are needed for score calculation of Transcription Factor Binding Sites (TFBS) because one position can have a big influence over TFBS score. Although only "A, C, G, T" characters are allowed for score calculation Morpheus gives the worst possible score (exact value depends of the matrix) if site contains any ambiguous character (IUPAC DNA code: A, C, G, T, U, R, Y, S, W, K, M, B, D, H, V, N, . , -). Log file will contain a list with sequences where this situation had been detected and it must be taken into account for correct interpretation of results.
Genomic information : If genomic information are available (chromosome, start position and sequence size) as in the case of ChIP-CHIP or ChIP-seq data, it can be indicated using "-" in sequence name as follow :
Name-chromosome-start position-size
For instance :

Occupancy result files have independent columns for genomic information (empty if not available).
You can download an example here.
LogInfo file : You can find in the LogInfo file information about the process as the number of sequences that have been identified, matrix data used or any problem in sequences/matrix recognition.
MATRIX
Matrix information can be found in transcription factor databases as JASPAR or directly generated from a DNA sequence alignment using tools as MEME. Here we describe the adequate way to write that information so that it can be correctly interpretedHere we describe the adequate way to write that information so that it can be correctly interpreted by Morpheus. In order to facilitate conversion, mPWM tool is available to generate Morpheus matrix format directly from a binding sites sequence alignment.
Morpheus matrix Conversion tool
This tool needs a file with binding sites sequence alignment either in fasta format or only aligned sequences. All sequences must have the same size, in other case the smaller size will be used. All IUPAC characters for DNA are allowed although only ACGT characters will be used for counting.
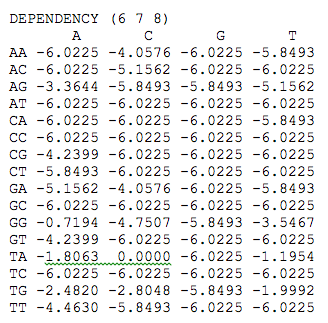
Optionally, if dependencies must be taken into account another file in txt format with dependencies positions will be necessary as well. In this file positions for each dependency must be indicated between squares brackets, one dependency per line. As for example :

User must select if matrix is symmetric or asymmetric and in the case of symmetric matrices, the corresponding symmetric dependencies will be generated even if they have not been indicated in the dependency information file. If the dependencies indicated in the example describes dependencies in a symmetric binding site of a total of 15 positions, program will take information of 4 dependencies :

File generated will contain matrix information in Morpheus format ready to use with any analysis tool in Morheus web.
Matrix Format
Format from MEME tool can be copied directly to generate de Matrix file (*.txt format).
The simplest format for a Matrix file is as follow :
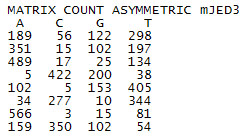
You can download an example here.
The first line specifies characteristics of the matrix separated by whitespaces, followed by information of nucleotide occurrence for each position (one position by line; in the example, a binding site of size 8 positions).
MATRIX --> Indicate that next data is the matrix data. ALL matrix files must start with this word.
Type of Data --> COUNT or FREQUENCY or SCORE
- COUNT --> Data is the occurrence number of each nucleotide (ACGT)
- FREQUENCY --> Data is the occurrence frequency of each nucleotide (ACGT)
- SCORE --> Data is the position weight matrix.
- If the data is in "COUNT" or "FREQUENCY" formats, transformation to weights data is done as follows:
- For each position --> Scorent(ACGT) =
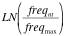
SYMMETRY --> ASYMMETRIC or SYMMETRIC
In some cases, due to structure of transcription factor or because it binds as homodimer, the matrix is palindromic, so that the scores of a sequence and of its reverse complement are the same. In those cases matrix has a SYMMETRIC structure.
If SYMMETRIC option is chosen but the matrix is asymmetric, scores will correspond only with one side (sense) estimation.
NAME --> The name of your matrix.
Default values : Only the term MATRIX and type of matrix are required for running the program. If one or both of the two others descriptive data (symmetry or name) are missing, the program will use default parameters.
- Symmetry --> ASYMMETRIC
- Name --> Unknown
LogInfo file : You can find in the LogInfo file information about matrix importation.
DEPENDENCY DATA
Position weight matrices assume that each base of the binding site contributes independently to the Transcription Factor/DNA affinity. However, there is evidence that interdependencies between positions exist and should be taken into account. Morpheus format allows using independent positions together with dependency in the needed positions.
For dependency data, a line with the word "DEPENDENCY" followed by the positions implicated, between brackets, precedes dependency data. This data is organized by alphabetical order for duplets (AA, AC, AG, ..., TG, TT) or triplets (AAA, AAC, AAG, ..., TTG, TTT). In order to facilitate data visualization the use of [A,C,G,T] characters are allowed, for example :
Data in independent matrix for positions implicated in dependencies are not taken into account for score calculation.
INPUT FILES
Score & Occupancy : These programs need a file with matrix information (Morpheus format) and a file with sequences (Fasta format).
ROC-AUC : This program needs two result files from Score (option best only) or Occupancy, one with the positive data and the other with the negative data.
OUTPUT FILES
Two types of files are generated from each program with TFBS information :
- File(s) in txt format with data organized in columns (score, position, TFBS sequence, etc.)
- Image file(s) with TFBS information (score profil, score distribution, ROC-AUC, etc.), using general parameters.
If you need other graphic representation for specific objectives, txt files contains all needed data and can be easily imported to other programs for graphical representation
We have realised that txt output files are not correctly visualized with some simple text processor. If you are not able to see information in organized columns try to use other text processor.