
Recursive Automatic Search of MOTif in 3D structures of PROteins
RASMOT-3D PRO: Supplementary informations
Associated publications
If you publish work in which you have used RASMOT-3D PRO, please cite the following publication:
Magis C, Gasparini D, Lecoq A, Le Du MH, Stura E, Charbonnier JB, Mourier G, Boulain JC, Pardo L, Caruana A, Joly A, Lefranc M, Masella M, Menez A, Cuniasse P., Structure-based secondary structure-independent approach to design protein ligands: Application to the design of Kv1.2 potassium channel blockers. 2006 Dec 20;128(50):16190-205 pubmed
Magis C, Gasparini D, Lecoq A, Le Du MH, Stura E, Charbonnier JB, Mourier G, Boulain JC, Pardo L, Caruana A, Joly A, Lefranc M, Masella M, Menez A, Cuniasse P., Structure-based secondary structure-independent approach to design protein ligands: Application to the design of Kv1.2 potassium channel blockers. 2006 Dec 20;128(50):16190-205 pubmed
Privacy policy
RASMOT-3D PRO policy is to respect and protect the privacy of our users. All collected informations (e-mail adress, uploaded structure files) are considered confidential.
We do not sell, trade, nor give away informations to other organizations. All uploaded informations are destroyed from our servers after 1 day.
We do not sell, trade, nor give away informations to other organizations. All uploaded informations are destroyed from our servers after 1 day.
Examples
To try the webserver, you can download the example compressed file containing PDB files of:
- different motifs :
- CD4 binding motif (Vita et al, 1999)
- Zinc finger motif
- for each motif:
- the target coordinates,
- the original complex,
- a structure in which the motif is found.
PDB file format
The Protein Data Bank (PDB) format provides a standard representation for macromolecular
structure data derived from X-ray diffraction and NMR studies.
Only ATOM records, which contains the atoms specifications, are read by RASMOT-3D PRO. For a complete description of the ATOM record format, see the Protein Data Bank Contents Guide .
Only ATOM records, which contains the atoms specifications, are read by RASMOT-3D PRO. For a complete description of the ATOM record format, see the Protein Data Bank Contents Guide .
Motif residues equivalence
RASMOT-3D principle is to search for residues exhibiting similar topology as the uploaded reference motif. This search can be restricted to residues with similar physical properties than their equivalent in the reference motif.
3 options are available :
3 options are available :
- all residues, i.e. no restriction : for each reference motif residue, all the residues are considered as potential equivalent in the compared structure. With this option, only topology is taken into account.
- identical residues, i.e. only same residue as in the reference motif is accepted as equivalent.
- same properties residues : for each reference motif residue, residues belonging to the same groups are considered as potential equivalent in the compared structure.
| acidic | Asp, Glu |
| basic | Lys, Arg, His |
| polar | Asn, Gln, Ser, Thr, Cys, His, Tyr |
| non polar | Val, Met, Ile, Leu, Trp, Phe, Ala, Gly, Pro |
| aromatic | Tyr, His, Phe, Trp |
| AsGlX | Asp, Glu, Asn, Gln |
delta-dist value
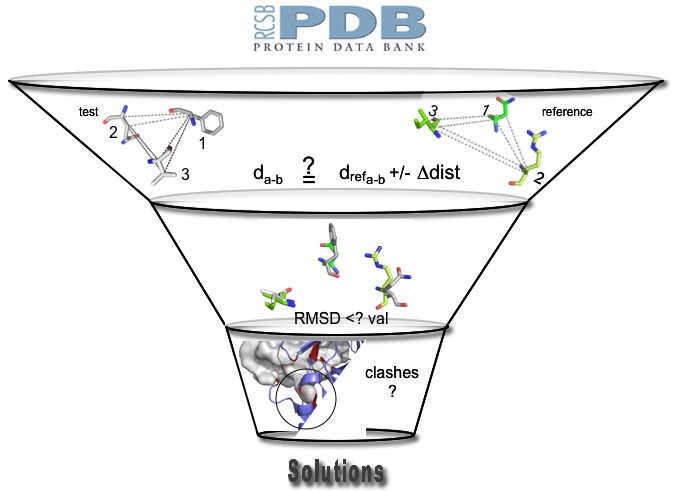 delta-dist value correspond to a maximum deviation criterion between interatomic (CA and CB atoms) distances describing the motif examined and the reference one. If at least one of these interatomic distances in the examined motif differs by more than delta-dist from the corresponding one in the reference motif, the motif is rejected.
delta-dist value correspond to a maximum deviation criterion between interatomic (CA and CB atoms) distances describing the motif examined and the reference one. If at least one of these interatomic distances in the examined motif differs by more than delta-dist from the corresponding one in the reference motif, the motif is rejected.This value has a great influence on the computing times. If it is to small, motifs satisfying all other criteria can be eliminated. If it is to arge, the number of motifs to examine with more time consumming filters grow exponentially. In practice, these value should be 1.5 to 2 times the RMSD value.
RMSD value
Root Mean Square Deviation is a standard measure of structural distance between coordinate sets.
The CA and CB atoms of the motif found are superimposed on the reference CA and CB atoms.
Then RMSD is calculated as the mean distance between the atoms of the identified motif and their
equivalents in the reference motif.
In RASMOT-3D PRO, RMSD have to be small enough to reproduce at best the reference motif topology but large enough to allow for minor deviations. In practice, RMSD value should range from 0.5 to 1.5 A.
In RASMOT-3D PRO, RMSD have to be small enough to reproduce at best the reference motif topology but large enough to allow for minor deviations. In practice, RMSD value should range from 0.5 to 1.5 A.
Steric filter
If you want to filter solutions making important steric clashes with a target (for binding motifs), you
have to provide a PDB file of the structure of the target placed correctly relative to the reference
motif.
Once the structure reproducing the motif topology (scaffold) is superimposed on the motif of reference, RASMOT-3D PRO counts its atoms that inter-penetrate target atoms. Inter-penetration is considered when the distance separating one atom of the scaffold and one atom of the target is below the sum of the corresponding atom radii. If the score associated to that number is to large, the structure is eliminated. The calculated score take into account the importance of the inter-penetration but also the distance to the backbone of the atoms making clashes (i.e. belong to the backbone, is located at the end of a flexible sidechain, etc...).
Once the structure reproducing the motif topology (scaffold) is superimposed on the motif of reference, RASMOT-3D PRO counts its atoms that inter-penetrate target atoms. Inter-penetration is considered when the distance separating one atom of the scaffold and one atom of the target is below the sum of the corresponding atom radii. If the score associated to that number is to large, the structure is eliminated. The calculated score take into account the importance of the inter-penetration but also the distance to the backbone of the atoms making clashes (i.e. belong to the backbone, is located at the end of a flexible sidechain, etc...).
Output file
Results are sorted according to the RMSD of the motif found from the reference one. For each
solution, are given :
Output file example :
- file : the name of the associated superimposed structure and the PyMol visualization files
- protein and chain : the name of the file from the PDB describing the structure and the corresponding chain in that file
- size : the number of residues of the protein
- model : if the structure has been obtained by NMR, several models can be present in the structure file, this field indicate the number of the model which fits the best to the reference motif
- rmsd : the RMSD calculated between CA and CB atoms of the motif found and the reference one
- motif : names and numbers of the residues constituting the motif found.
Output file example :
Found 852 solutions (250 detailed)
file protein chain size model rmsd motif
pdb2dl5_00 2dl5 A 78 19 0.31 {TYR 22 , GLN 26 , GLU 29 , TRP 49 }
pdb1uhc_00 1uhc A 79 3 0.31 {PHE 22 , ASN 26 , GLU 29 , TRP 52 }
pdb1yn8_01 1yn8 A 54 1 0.38 {PHE 10 , ASN 14 , GLU 17 , TRP 36 }
pdb2co5_00 2co5 B 94 1 0.39 {GLU 80 , ARG 8 , TYR 11 , SER 53 }
pdb1k0x_00 1k0x A 108 10 0.39 {TYR 31 , ASP 35 , PHE 38 , PHE 60 }
pdb1wxu_00 1wxu A 93 1 0.40 {PHE 26 , SER 30 , GLU 33 , TRP 56 }
pdb1z9z_00 1z9z A 59 1 0.41 {PHE 11 , SER 15 , GLU 18 , TRP 38 }
pdb1w2q_00 1w2q A 127 6 0.41 {PHE 111 , MET 67 , THR 70 , ALA 77 }
pdb1x6b_00 1x6b A 79 17 0.41 {PHE 26 , GLN 30 , GLU 33 , TRP 51 }
pdb1j3t_00 1j3t A 74 17 0.42 {TRP 19 , LYS 23 , HIS 26 , TRP 44 }
|
Visualization of the results
a) Local visualization with PyMol
You can download an archive containing the output, the superimposed structures and the PyMol visualization files.
Unzip the archive. For each solution, two files are available : a PDB file of the superimposed structure and a PML file, which is a visualization script file forPyMol (a free
version of the software can be downloaded at http://delsci.com/rel/099/).
To visualize a solution :
NB: the solution structure PDB files can be viewed with all the PDB visualization software but provided visualization scripts only works with PyMol.
b) Online visualization with Jmol
Jmol is a java molecular viewer for 3D structures. Applet is integrated into webpages and does not require any plugin installation. Jmol only requires Java installation (http://www.java.com/) and operates on a wide variety of platforms.
On the result page, just click on the file name of the choosen result.
You can download an archive containing the output, the superimposed structures and the PyMol visualization files.
Unzip the archive. For each solution, two files are available : a PDB file of the superimposed structure and a PML file, which is a visualization script file for
To visualize a solution :
- launch PyMol
- in the File menu, click on Run...
- choose the PML file you want to run, then click on Open
NB: the solution structure PDB files can be viewed with all the PDB visualization software but provided visualization scripts only works with PyMol.
b) Online visualization with Jmol
Jmol is a java molecular viewer for 3D structures. Applet is integrated into webpages and does not require any plugin installation. Jmol only requires Java installation (http://www.java.com/) and operates on a wide variety of platforms.
On the result page, just click on the file name of the choosen result.
