BRIDGED
Disulfide bond prediction
BRIDGED: Supplementary informations
About the input :
BridgeD requires a regular-formatted PDB file: only the ATOM lines will be considered in the computation. A structure check is performed prior to computation but spurious cases (errors in PDB) are still possible.
The search for putative disulfide bonds is restricted to a user-given PDB chain. If the PDB does not contain a chain Id, then leave the box empty.
To avoid any bias from existent disulfide bonds in a three-dimensional structure, any Cys residues could be replaced by alanines beforehand; to do so check the corresponding box.
The resolution of a run concerns the clustering radius used to minimized side chains around the modelled disulfide bond (low=5.45, med=5.95, high=6.45). The published method uses the low value [1].
About the results:
A zip file is sent as a result. It contains a file name: ssbond_XXXX.out where XXXX is an integer number. It also contains a file name: ssbond_XXXX.log which is the raw output of bridgeD; the ssbond_XXXX.out file is extracted from the .log file.
A directory named PDBOUTS is included and contains all the modelled disulfide bonds in the template PDB
Important information:
In the testing of this disulfide bond prediction method [1] we found that using multiple PDB templates improved the chance of finding reported experimental engineered disulfide bonds: accuracy in prediction rose from 84% to 93%. Because the output data is a text file, it is easy to run multiple PDB templates and then concatenate lines. A simple sorting script will remove duplicated lines.
About the method:
In the initial stage, the BridgeD program screens paired residues in a protein for collecting plausible disulfide bond sites by the distance criterion, Dcb-cb 5.05 Å. By default, the chosen paired residues are separated by at least two residues in the protein sequence (contact the authors if you need a different value). A clustering procedure allows cross-pairing to occur between different pairs from initial screening, if the Cb-Cb distance between the cross-paired residues is within the clustering radius (5.55 Å). The groups of optimized side-chains were chosen by their Cb atoms within 5.45 Å of the Cb atoms of the mutated residues. The side-chain minimization algorithm combined both discrete (rotamer-based) and continuous energy minimization to obtain the best side-chain conformation for the optimized residues [2].
It should be noticed that no all-atom minimization is performed at the end of the run. In the accompanying paper, a final refinement stage was performed by X-PLOR. However, the quality of prediction was not affected by the minimization step since the final total energy of the model was not used as a criterion to characterize disulfide bond predictions.
To automatically derive the bonding state of the two engineered cysteines from structure optimization, we introduced a weighing factor, lambda, into the scoring function, E, defined as
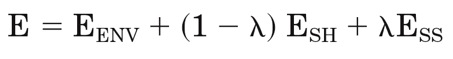
where EENV represents the nonbonded potential energy of the protein environment interacting with other optimized side-chain residues than the engineered pair. ESH represents
the nonbonded interactions of the reduced Cys residues with surroundings and their mutual interaction; ESS represents that of the oxidized Cys-Cys pair and their bonding energy, Ebond which is solely determined by DSg-Sg and Css.
Analogous to the Boltzmann distribution law, the weighing factor is approximated as
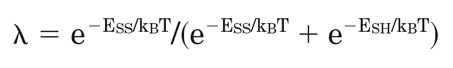
where kBT equals 0.59, a product of the Boltzmann constant and the temperature at 298 K. If Cys-Cys are predicted as in the oxidized state, then lambda=1, whereas reduced Cys-Cys are indicated by lambda=0.
About disulfide bond stability:
The stability of a reported engineered disulfide bond may be inferred by its mutational cost and the weighing factor lambda. If an engineered disulfide bond causes great conformational disturbances, the mutation may bring the protein into an unstable state. By calculating DE, BridgeD highlights the putative engineered disulfide bond that may destabilize the protein, or at least distort the present backbone conformation. By calculating lambda , one may determine which oxido-reduction state is preferred for the paired Cys residues in the present structural framework.
References
[1] Pellequer J-L, Chen S-wW. approach to modeling engineered disulfide bonds. Proteins 2006; 65:192-202.
[2] Chen S-wW, Pellequer JL. Identification of functionally important residues in proteins using comparative models. Curr Med Chem 2004;11:595–605.
